
-
中文 | EN
中文 | EN
发布时间:2020-12-14 00:00:00.0
细胞死亡受体相互作用的丝氨酸/苏氨酸蛋白激酶1(RIPK1)是调控死亡受体和模式识别受体信号通路的重要蛋白。RIPK1含有三个重要的结构域,N端的蛋白激酶结构域,含有RHIM(RIP Homotypic Interaction Motif)的中间结构域和C端的死亡结构域(death domain)。在肿瘤坏死因子a(TNFa)刺激的条件下,RIPK1激酶通过激活RIPK1依赖的细胞凋亡和坏死发挥着促进细胞死亡和促进炎症的作用1, 2。但是,已有的研究表明RIPK1不依赖其激酶活性的支架功能发挥着促进存活的作用。RIPK1缺陷在一些罕见的人类病例中导致免疫缺陷和肠炎3。RIPK1敲除的小鼠在出生时死于系统性炎症,RIPK3和caspase-8双敲除可以挽救小鼠的围产期死亡,Toll-like receptor 3/4(TLR3/4)信号通路的关键调控因子Myd88或者TRIF敲除可以缓解该表型4, 5。但是,是什么机制调控RIPK1发挥两种不同的作用尚不清楚。RIPK1的死亡结构域(DD)介导了其在TNF受体复合物中的招募6,并且RIPK1通过DD形成二聚体促进其激酶活性的激活7。RIPK1复杂的泛素化修饰以及与其他含有DD的蛋白包括TRADD,FADD和TNFR1的相互作用精密地调控RIPK1激酶的激活8。但是,是什么调控了RIPK1与其他含有DD的蛋白的相互作用以及不同的刺激条件下RIPK1泛素化如何调控RIPK1激酶的激活仍然不清楚。在TLR通路中,LPS刺激促进RIPK1/RIPK3/FADD/caspase-8复合物形成,激活RIPK3参与的caspase-1激活及IL-1b的成熟,从而介导炎症反应9, 10。但是,RIPK1在其中的支架蛋白如何抑制RIPK3的激活及其下游caspase-1激活及IL-1b的剪切成熟尚不清楚。
2020年12月11日,中国科学院上海有机化学研究所生物与化学交叉研究中心袁钧瑛课题组在Nature Communications 上在线发表文章Ubiquitination of RIPK1regulates its activation mediated by TNFR1 and TLR signaling in distinct manner。本工作通过系统地突变RIPK1泛素化赖氨酸位点,筛选出人源RIPK1死亡结构域的627位赖氨酸以及其同源的鼠RIPK1612位赖氨酸对于介导RIPK1泛素化,RIPK1同源二聚及异源结合TNFR1,TRADD和FADD非常重要。通过质谱分析RIPK1肽段的泛素化修饰,我们发现人RIPK1 K627及鼠RIPK1 K612的泛素化调控RIPK1的整体泛素化模式,促进RIPK1 N端蛋白激酶结构域和中间结构域泛素化水平,抑制死亡结构域的泛素化。RIPK1 K612R突变抑制了RIPK1-DD与RIPK1-DD的同源二聚,以及RIPK1-DD与TNFR1-DD,TRADD-DD 和FADD-DD的异源二聚。通过建立和分析RIPK K612R突变小鼠,我们发现K612R突变抑制TNFa刺激引起的RIPK1的激活,从而抑制RIPK1依赖的细胞凋亡和坏死。然而,RIPK1K612R/K612R骨髓来源的巨噬细胞在TLR3/4通路中促进RIPK3的激活及RIPK3/RIPK1/MLKL复合物的形成,促进Caspase-1的剪切,IL-1b的释放以及炎症反应。RIPK1K612R/K612R小鼠随着年龄的增长出现自发性的结肠炎和脾肿大,表现为结肠免疫细胞浸润和炎症因子表达增加,以及脾和骨髓中CD11b+GR-1+的粒细胞增多。喂食抗生素抑制小鼠的结肠炎和脾肿大,RIPK3敲除可以挽救脾肿大以及脾和骨髓中CD11b+GR-1+的粒细胞的增加及部分挽救结肠炎。我们发现K612R RIPK1与FADD的相互结合缺陷是导致RIPK1K612R/K612R细胞在TLR3/4通路中对于坏死异常敏感和caspase-1激活的重要原因。综上所述,人源RIPK1 K627及鼠源RIPK1 K612的泛素化在TNFR1和TLRs信号通路中起到了不同的调控功能。本工作突显了RIPK1 DD和hRIPK1 K627/mRIPK1 K612泛素化对于维持肠道稳定性的重要作用,揭示了细胞的不同死亡形式包括凋亡,坏死和caspase-1激活之间进行交互作用的分子机制,为研究人类肠炎等免疫性疾病提供了良好的动物模型和理论基础。
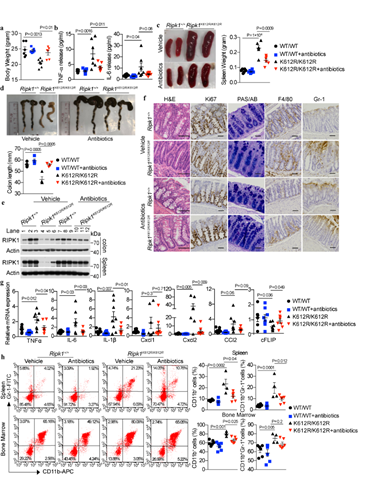
图1. 抗生素抑制RIPK1 K612R突变小鼠的自发结肠炎和脾肿大

图2.RIPK1 K612R突变调控TNFR1 和TLR3/4信号通路的模式图
该论文的共同通讯作者为袁钧瑛研究员和单冰副研究员,第一作者是中科院上海有机化学研究所生物与化学交叉研究中心的李兴彦博士。
1. Mifflin,L., Ofengeim, D. & Yuan, J. Receptor-interacting protein kinase 1 (RIPK1)as a therapeutic target. Nature reviews.Drug discovery 19, 553-571(2020).
2. Yuan,J., Amin, P. & Ofengeim, D. Necroptosis and RIPK1-mediatedneuroinflammation in CNS diseases. Naturereviews. Neuroscience 20, 19-33(2019).
3. Cuchet-Lourenco,D. et al. Biallelic RIPK1 mutationsin humans cause severe immunodeficiency, arthritis, and intestinalinflammation. Science 361, 810-813 (2018).
4. Dillon,C.P. et al. RIPK1 blocks earlypostnatal lethality mediated by caspase-8 and RIPK3. Cell 157, 1189-1202 (2014).
5. Newton,K. et al. RIPK1 inhibits ZBP1-drivennecroptosis during development. Nature540, 129-133 (2016).
6. Hsu,H., Huang, J., Shu, H.B., Baichwal, V. & Goeddel, D.V. TNF-dependentrecruitment of the protein kinase RIP to the TNF receptor-1 signaling complex. Immunity 4, 387-396 (1996).
7. Meng,H. et al. Death-domaindimerization-mediated activation of RIPK1 controls necroptosis andRIPK1-dependent apoptosis. Proceedings ofthe National Academy of Sciences of the United States of America 115, E2001-E2009 (2018).
8. Shan,B., Pan, H., Najafov, A. & Yuan, J. Necroptosis in development anddiseases. Genes & development 32, 327-340 (2018).
9. Schroder,K. & Tschopp, J. The inflammasomes. Cell140, 821-832 (2010).
10. Moriwaki,K., Bertin, J., Gough, P.J. & Chan, F.K. A RIPK3-caspase 8 complex mediatesatypical pro-IL-1beta processing. JImmunol 194, 1938-1944 (2015).